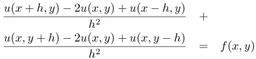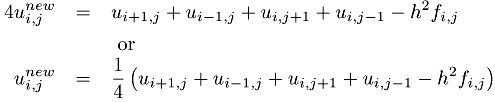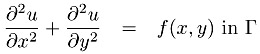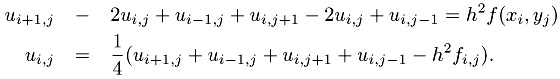Chapter 7: An Introduction to Writing Parallel Programs for Clusters
Overview
Ewing Lusk, William Gropp, and Ralph Butler
There are two common kinds of parallelism. The first, the master-worker approach, is the simplest and easiest to implement. It relies on being able to break the computation into independent tasks. A master then coordinates the solution of these independent tasks by worker processes. This kind of parallelism is discussed in detail in this Chapter, starting with
Section 7.1. This part of the chapter has three goals:
-
To present some of the ways parallelism can be introduced into an application.
-
To describe how to express parallelism using functions built into the operating system. Depending on your target application, this information may be all you need, and we will show how to access these functions from several different application-development languages.
-
To provide some realistic examples of applications illustrating this approach. We include string matching with applications to computational biology.
The first task in creating a parallel program is to express concurrent work.
Section 7.1 then focuses on task parallelism.
Section 7.2 describes the use of Linux system calls for task parallelism.
Section 7.4 then outlines an example from computational biology to illustrate the use of task parallelism to carry out a large computation.
The second kind of parallelism is for computations that cannot (or cannot easily) be broken into independent tasks. In this kind of parallelism, the computation is broken down into communicating, interdependent tasks. One example of this kind of parallelism was introduced in
Section 1.3.6. These parallel programs are more difficult to write, and a number of programming models have been developed to support this kind of parallel program. The most common, and the one most appropriate for Beowulf clusters, is
message passing. The two most common message-passing systems are MPI (Message Passing Interface) and PVM (Parallel Virtual Machine), covered in Chapters 8–11. In this chapter,
Section 7.5 introduces some of the techniques used in dividing programs into communicating, interdependent parts. Expressing these ideas as programs is left for the following chapters.
7.1 Creating Task ParallelismThe essence of task parallelism is that the task to be accomplished can be executed in parallel. Since we assume that the tasks are not completely independent (otherwise they are just a collection of ordinary sequential jobs), some sort of coordinating mechanism must exist. We will call this process the manager, and the processes that carry out the subtasks the workers. (The manager could even be the human user, who manages the worker processes "by hand," but we will assume that the manager is a single program that the user causes to be started.) Manager/worker algorithms and execution mechanisms have many variations, which we survey in the
next section; but as we use the term, "task parallelism" always involves the following steps.
-
Divide the task into independent or nearly independent subtasks. By "independent" we mean that while communication of some sort occurs between the manager and the workers, there is no direct communication between any two workers.
-
Start the workers. We assume that each worker is represented by an operating system process. In
Section 7.2 we will describe Unix processes and how to start them. (Use of threads for workers is atypical for a Beowulf cluster and will not be described.)
-
Communicate subtask specifications from the manager to the workers.
-
Communicate results from the workers to the manager.
-
Ensure that all results have been collected and that the workers have been shut down.
7.1.1 Variations on Task Parallelism
The scheme just described had many variations; we will discuss a few of them here, and then in the following section we will illustrate some of these with concrete examples. The variations involve the scheduling algorithm by which the manager assigns subtasks to the workers, the ways in which the worker processes are started and managed, and the communication mechanism between manager and workers.
Variations in How Work Is Assigned
For an efficient manager/worker parallel program, the workers should be kept working as much of the total time as possible. If the total work to be done can be easily divided into arbitrarily sized subtasks, then the scheduling job is easy: if there are n workers, then divide the work up into n pieces, each of which will take the same amount of time, and give one piece to each worker. This is called static scheduling.
Although sometimes such scheduling can be done, breaking up the total amount of work into subtasks typically results in subtasks of widely differing sizes, more subtasks than there are workers, or both. In all of these cases, the manager must organize the assignment of work to workers more carefully in order to keep the workers working. If some workers are idle when there is still more work to do, a load balancing problem occurs. Fortunately the general manager/worker algorithms can be used to overcome this problem when there are substantially more subtasks than there are workers. The idea is for the manager to make an initial assignment of subtasks to workers and then wait for subtask completion by any worker. At that point the worker can be assigned another subtask. In this way the master does not need to know ahead of time how much time each subtask will take; it just keeps all the workers as busy as possible.
Figure 7.1 shows a high-level framework for the manager and worker in a manager/worker system. In this example, n processes (workers) are started and then each process is sent the data for each task to perform. New processes are started once rather than for each task, because starting a new process is often a time-consuming operation.
We note a few points about this algorithm.
-
A load balancing problem will inevitably occur near the end of the job, as some workers become idle but there is no more work to be assigned, because all the work that is not done is being worked on by other workers.
-
To make this period of load imbalance as small as possible, it is a good idea to make the subtasks small. If the manager can estimate the sizes of the subtasks, then it should assign the larger tasks first and the smaller ones near the end.
-
If the subtasks are too small, then the overhead of communication between manager and worker will take up too much of the total time.
Therefore one needs to give some thought to just exactly how to divide up the work. A technique that is sometimes used is to further subdivide the work units during the computation. In some algorithms, the workers subdivide their own tasks and return the new subsubtasks to the manager for redistribution to the other workers. An example is the Mandelbrot program described in
Chapter 5 of [48].
Variations in Implementation Mechanisms
Processes can be started in a variety of ways, including shell commands, Unix system calls, remote shell commands of different kinds, parallel process management systems, and the use of daemons of various kinds. Even Web browsers can be used to launch remote tasks. We will discuss process startup in
Section 7.2, after we have established a deeper understanding of operating system processes.
Similarly, the communication between manager and worker can be carried out in many ways. One way is to use the file system as a communication device. This is particularly convenient if all of the workers have access to the same file system. (See
Chapter 19 for a discussion of shared file systems.) This mechanism is often used when the manager is programmed as a shell script.
A more flexible and powerful approach to communication among processes uses sockets. Sockets and how to use them in several programming languages are covered in
Section 7.2.5. The use of higher-level communication libraries (MPI and PVM) is covered in later chapters.
7.2 Operating System Support for ParallelismAlthough parallel programs can be quite complex, many applications can be made parallel in a simple way to take advantage of the power of Beowulf clusters. In this section we describe how to write simple programs using features of the Linux operating system that you are probably already familiar with. We begin with a discussion of processes themselves (the primary unit of parallelism) and the ways they can be created in Unix environments such as Linux. A good reference on this material is
[111].
7.2.1 Programs and Processes
First we review terminology. A program is a set of computer instructions. A computer fetches from its memory the instruction at the address contained in its program counter and executing that instruction. Execution of the instruction sets the program counter for the next instruction. This is the basic von Neumann model of computation. A process consists of a program, an area of computer memory called an address space, and a program counter. (If there are multiple program counters for a single address space, the process is called a multithreaded process.) Processes are isolated from one another in the sense that no single instruction from the program associated with one process can access the address space of another process. Data can be moved from the address space of one process to that of another process by methods that we will describe in this and succeeding chapters. For the sake of simplicity, we will discuss single-threaded processes here, so we may think of a process as an (address space, program, program counter) triple.
7.2.2 Local Processes
Where do processes come from? In Unix-based operating systems such as Linux, new processes are created by the fork system call. This is an efficient and lightweight mechanism that duplicates the process by copying the address space and creating a new process with the same program. The only difference between the process that executed the fork (called the parent process) and the new process (called the child process) is that the fork call returns 0 in the child and the process id in the parent. Based on this different return code from fork, the parent and child processes, now executing independently, can do different things.
One thing the child process often does is an exec system call. This call changes the program for the process, sets the program counter to the beginning of the program, and reinitializes the address space. The fork-exec combination, therefore, is the mechanism by a process create a new, completely different one. The new process is executing on the same machine and competing for CPU cycles with the original process through the process scheduler in the machine's operating system.
You have experienced this mechanism many times. When you are logged into a Unix system, you are interacting with a shell, which is just a normal Unix process that prompts you, reads your input commands, and processes them. The default program for this process is /bin/bash; but depending on the shell specified for your user name in '/etc/passwd', you may be using another shell. Whenever you run a Unix command, such as grep, the shell forks and execs the program associated with the command. The command ps shows you all the processes you are running under the current shell, including the ps process itself (strictly speaking, the process executing the ps program).
Normally, when you execute a command from the shell, the shell process waits for the child process to complete before prompting you for another command, so that only one process of yours at a time is actually executing. By "executing" we mean that it is in the list of processes that the operating system will schedule for execution according to its time-slicing algorithm. If your machine has ony one CPU, of course only one instruction from one process can be executing at a time. By time-slicing the CPU among processes, however, the illusion of simultaneously executing process on a single machine, even a single CPU, is presented.
The easiest way to cause multiple processes to be scheduled for execution at the same time is to append the '&' character to a command that you execute in the shell. When you do this, the shell starts the new process (using the fork-exec mechanism) but then immediately prompts for another command without waiting for the new one to complete. This is called "running a process in the background." Multiple background processes can be executing at the same time. This situation provides us with our first example of parallel processes.
To determine whether a file contains a specific string, you can use the Unix command grep. To look in a directory containing mail files in order to find a message about the Boyer-Moore string-matching algorithm, you can cd to that directory and do
If your mail is divided into directories by year, you can consider search all those directories in parallel. You can use background processes to do this search in a shell script:
and invoke this with Boyer as an argument.
This simple parallel program matches our definition of a manager/worker algorithm, in which the master process executes this script and the worker processes execute grep. We can compare its properties with the list in
Section 7.1:
-
The subtasks, each of which is to run grep over all the files in one directory, are independent.
-
The workers are started by this shell script, which acts as the master.
-
The subtask specifications (arguments to grep) are communicated to the workers on their respective command lines.
-
The results are written to the file system, one result file in each directory.
-
The wait causes the shell script to wait for all background processes to finish, so that the results can be collected by the manager (using cat) into one place.
One can make a few further observations about this example:
-
The first line of the script tells the system which program to use to interpret the script. Here we have used the default shell for Linux systems, called bash. Other shells may be installed on your system, such as csh, tcsh, or zsh. Each of these has a slightly different syntax and different advanced features, but for the most part they provide the same basic functionality.
-
We could have made the size of the subtask smaller by running each invocation of grep on a single file. This would have led to more parallelism, but it is of dubious value on a single machine, and we would have been creating potentially thousands of processes at once.
-
We could time this script by putting date commands at the beginning and end, or by running it under the shell's time command:
where grepmail is the name of this script and boyer is the argument.
7.2.3 Remote Processes
Recall that the way a process is created on a Unix system is with the fork mechanism. Only one process is not forked by another process, namely the single init process that is the root of the tree of all processes running at any one time.
Thus, if we want to create a new process on another machine, we must contact some existing process and cause it to fork our new process for us. There are many ways to do this, but all of them use this same basic mechanism. They differ only in which program they contact to make a fork request to. The contact is usually made over a TCP socket. We describe sockets in detail in
Section 7.2.5.
rsh
The rsh command contacts the rshd process if it is running on the remote machine and asks it to execute a program or script. To see the contents of the '/tmp' directory on the machine foo.bar.edu, you would do
The standard input and output of the remote command are routed through the standard input and output of the rsh command, so that the output of the ls comes back to the user on the local machine.
Chapter 5 describes how to set up rsh on your cluster.
ssh
The ssh (secure shell) program behaves much like rsh but has a more secure authentication mechanism based on public key encryption and encrypts all traffic between the local and remote host. It is now the most commonly used mechanism for starting remote processes. Nevertheless, rsh is substantially faster than ssh, and is used when security is not a critical issue. A common example of this situation occurs when the cluster is behind a firewall and rsh is enabled just within the cluster. Setting up ssh is described in
Chapter 5, and a book on ssh has recently appeared
[11].
Here is a simple example. Suppose that we have a file called 'hosts' with the names of all the hosts in our cluster. We want to run a command (in parallel) on all those hosts. We can do so with a simple shell script as follows:
If everything is working correctly and ssh has been configured so that it does not require a password on every invocation, then we should get back the names of the hosts in our cluster, although not necessarily in the same order as they appear in the file.
(What is that -x doing there? In this example, since the remotely executed program (hostname) does not use any X windowing facilities, we turn off X forwarding by using the -x option. To run a program that does use X, the X option must be turned on by the sshd server at each remote machine and the user should set the DISPLAY environment variable. Then, the connection to the X display is automatically forwarded in such a way that any X programs started from the shell will go through the encrypted channel, and the connection to the real X server will be made from the local machine. We note that if you run several X programs at several different hosts, they will each create a file named '.Xauthority' in your home directory on each of the machines. If the machines all have the same home directory, for example mounted via NFS, the '.Xauthority' files will conflict with each other.)
Other Process Managers
Programs such as the ones rsh and ssh contact to fork processes on their behalf are often called daemons. These processes are started when the system is booted and run forever, waiting for connections. You can see whether the ssh daemon is installed and running on a particular host by logging into that host and doing ps auxw | grep sshd. Other daemons, either run as root by the system or run by a particular user, can be used to start processes. Two examples are the daemons used to start processes in resource managers and the mpd's that can be used to start MPI jobs quickly (see
Chapter 8).
7.2.4 Files
Having discussed how processes are started, we next tunr to the topic of remote files, files that are local to a remote machine. Often we need to move files from one host to another, to prepare for remote execution, to communicate results, or even to notify remote processes of events.
Moving files is not always necessary, of course. On some clusters, certain file systems are accessible on all the hosts through a system like NFS (Network File System) or PVFS (Parallel Virtual File System). (
Chapter 19 describes PVFS in detail.) However, direct remote access can sometimes be slower than local access. In this section we discuss some mechanisms for moving files from one host to another, on the assumption that the programs and at least some of the files they use are desired to be staged to a local file system on each host, such as '/tmp'.
rcp
The simplest mechanism is the remote copy command rcp. It has the same syntax as the standard local file copy command cp but can accept user name and host information from the file name arguments. For example,
copies a local file to a specific location on the host specified by the prefix before the ':'. A remote user can also be added:
The rcp command uses the same authentication mechanism as rsh does, so it will either ask for a password or not depending on how rsh has been set up. Indeed, rcp can be thought of as a companion program to rsh. The rcp command can handle "third party" transfers, in which neither the source nor destination file is on the local machine.
scp
Just as ssh is replacing rsh for security reasons, scp is replacing rcp. The scp command is the ssh version of rcp and has a number of other convenient features, such as a progress indicator, which is handy when large files are being transferred.
The syntax of scp is similar to that for rcp. For example,
will log in to machine fronk.cs.jx.edu as user jones (prompting for a password for jones if necessary) and then copy the file 'bazz' in user jones's home directory to the file 'bazz' in the current directory on the local machine.
ftp and sftp
Both ftp and sftp are interactive programs, usually used to browse directories and transfer files from "very" remote hosts rather than within a cluster. If you are not already familiar with ftp, the man page will teach you how to work this basic program. The sftp program is the more secure, ssh-based version of ftp.
rdist
One can use rdist to maintain identical copies of a set of files across a set of hosts. A flexible 'distfile' controls exactly what files are updated. This is a useful utility when one wants to update a master copy and then have the changes reflected in local copies on other hosts in a cluster. Either rsh-style (the default) or ssh-style security can be specified.
rsync
An efficient replacement for rcp is rsync, particularly when an earlier version of a file or directory to be copied already exists on the remote machine. The idea is to detect the differences between the files and then just transfer the differences over the network. This is especially effective for backing up large directory trees; the whole directory is specified in the command, but only (portions of) the changed files are actually copied.
7.2.5 Interprocess Communication with Sockets
The most common and flexible way for two processes on different hosts in a cluster to communicate is through sockets. A socket between two processes is a bidirectional channel that is accessed by the processes using the same read and write functions that processes use for file I/O. In this section we show how a process connects to another process, establishing a socket, and then uses it for communication. An excellent reference for the deep topic of sockets and TCP/IP in general is
[111]. Here we just scratch the surface, but the examples we present here should enable you to write some useful programs using sockets. Since sockets are typically accessed from programming and scripting languages, we give examples in C, Perl, and Python, all of which are common languages for programming clusters.
Although once a socket is established, it is symmetric in the sense that communication is bidirectional, the initial setup process is asymmetric: one process connects; the other one "listens" for a connection and then accepts it. Because this situation occurs in many client/server applications, we call the process that waits for a connection the server and the process that connects to it the client, even though they may play different roles after the socket has been established.
We present essentially the same example in three languages. In the example, the server runs forever in the background, waiting for a socket connection. It advertises its location by announcing its host and "port" (more on ports below), on which it can be contacted. Then any client program that knows the host and port can set up a connection with the server. In our simple example, when the server gets the connection request, it accepts the request, reads and processes the message that the client sends it, and then sends a reply.
Client and Server in C
The server is shown in
Figure 7.2. Let us walk through this example, which may appear more complex than it really is. Most of the complexity surrounds the sockaddr_in data structure, which is used for two-way communication with the kernel.
First, we acquire a socket with the socket system call. Note that we use the word "socket" both for the connection between the two processes, as we have used it up to now, and for a single "end" of the socket as it appears inside a program, as here. Here a socket is a small integer, a file descriptor just like the ones used to represent open files. Our call creates an Internet (AF_INET) stream (SOCK_STREAM) socket, which is how one specifies a TCP socket. (The third argument is relevant only to "raw" sockets, which we are not interested in here. It is usually set to zero.) This is our "listening socket," on which we will receive connection requests. We then initialize the sockaddr_in data structure, setting its field sin_port to 0 to indicate that we want the system to select a port for us. A port is an operating system resource that can be made visible to other hosts on the network. We bind our listening socket to this port with the bind system call and notify the kernel that we wish it to accept incoming connections from clients on this port with the listen call. (The second argument to listen is the number of queued connection requests we want the kernel to maintain for us. In most Unix systems this will be 5.) At this point clients can connect to this port but not yet to our actual server process. Also, at this point no one knows what port we have been assigned.
We now publish the address of the port on which we can be contacted. Many standard daemons listen on "well known" ports, but we have not asked for a specific port, so our listening socket has been assigned a port number that no one yet knows. We ourselves find out what it is with the getsockname system call and, in this case, just print it on stdout.
At this point we enter an infinite loop, waiting for connections. The accept system call blocks until there is a connection request from a client. Then it returns a new socket on which we are connected to the client, so that it can continue listening on the original socket. Our server simply reads some data from the client on the new socket (talk_socket), echoes it back to the client, closes the new socket, and goes back to listening for another connection.
This example is extremely simple. We have not checked for failures of any kind (by checking the return codes from our system calls), and of course our server does not provide much service. However, this example does illustrate how to code a common sequence of system calls (the socket-bind-listen sequence) that is used in nearly all socket setup code.
The corresponding client is shown in
Figure 7.3. In order to connect to the server, it must know the name of the host where the server is running and the number of the port on which it is listening. We supply these here as command-line arguments.
Again we acquire a socket with the socket system call. We then fill in the sockaddr_in structure with the host and port (first calling gethostbyname to fill in the hostent structure needed to be placed in sin). Next we call connect to create the socket. When connect returns, the accept has taken place in the server, and we can write to and read from the socket as a way of communicating with the server. Here we send a message and read a response, which we print.
Client and Server in Python
Python is an object-oriented scripting language. Implementations exist for Unix and Windows; see
www.python.org for details. It provides an extensive set of modules for interfacing with the operating system. One interesting feature of Python is that the block structure of the code is given by the indentation of the code, rather than explicit "begin"/ "end" or enclosing braces.
Much of the complexity of dealing with sockets has to do with properly managing the sockaddr data structure. Higher-level languages like Python and Perl make socket programming more convenient by hiding this data structure. A number of good books on Python exist that include details of the socket module; see, for example,
[14] and [70]. Python uses an exception handling model (not illustrated here) for error conditions, leading to very clean code that does not ignore errors. The Python version of the server code is shown in
Figure 7.4. Here we use the "well-known port" approach: rather than ask for a port, we specify the one we want to use. One can see the same socket-bind-listen sequence as in the C example, where now a socket object (s) is returned by the socket call and bind, listen, and accept are methods belonging to the socket object. The accept method returns two objects, a socket (conn) and information (addr) on the host and port on the other (connecting) end of the socket. The methods send and recv are methods on the socket object conn, and so this server accomplishes the same thing as the one in
Figure 7.2.
The Python code for the corresponding client is shown in
Figure 7.5. It has simply hard-coded the well-known location of the server.
Client and Server in Perl
Perl
[124] is a powerful and popular scripting language. Versions exist for Unix and for Windows; see
www.perl.com for more information. Perl provides a powerful set of string matching and manipulation operations, combined with access to many of the fundamental system calls. The man page perlipc has samples of clients and servers that use sockets for communication.
The code for a "time server" in Perl is shown in
Figure 7.6. It follows the same pattern as our other servers. The code for the corresponding client is shown in
Figure 7.7.
7.2.6 Managing Multiple Sockets with Select
So far our example socket code has involved only one socket open by the server at a time (not counting the listening socket). Further, the connections have been short lived: after accepting a connection request, the server handled that request and then terminated the connection. This is a typical pattern for a classical server but may not be efficient for manager/worker algorithms in which we might want to keep the connections to the workers open rather than reestablish them each time. Unlike the clients in the examples above, the workers are persistent, so it makes sense to make their connections persistent as well.
What is needed by the manager in this case is a mechanism to wait for communication from any of a set of workers simultaneously. Unix provides this capability with the select system call. The use of select allows a process to block, waiting for a change of state on any of a set of sockets. It then "wakes up" the process and presents it with a list of sockets on which there is activity, such as a connection request or a message to be read. We will not cover all of the many aspects of select here, but the code in
Figure 7.8 illustrates the features most needed for manager/worker algorithms. For compactness, we show this in Python. A C version would have the same logic. See the select man page or
[111] for the details of how to use select in C. It is also available, of course, in Perl.
The first part of the code in
Figure 7.8 is familiar. We acquire a socket, bind it to a port, and listen on it. Then, instead of doing an accept on this socket directly, we put it into a list (sockets). Initially it is the only member of this list, but eventually the list will grow. Then we call select. The arguments to select are three lists of sockets we are interested in for reading, writing, or other events. The select call blocks until activity occurs on one of the sockets we have given to it. When select returns, it returns three lists, each a sublist of the corresponding input lists. Each of the returned sockets has changed state, and one can take some action on it with the knowledge that the action will not block.
In our case, we loop through the returned sockets, which are now active. We process activity on the listening socket by accepting the connection request and then adding the new connection to the list of sockets we are interested in. Otherwise we read and print the message that the client has sent us. If our read attempt yields an empty message, we interpret this as meaning that the worker has closed its end of the socket (or exited, which will close the socket), and we remove this socket from the list.
We can test this server with the client in
Figure 7.9.
 |
#!/usr/bin/env python
from sys import argv, stdin
from socket import socket, AF_INET, SOCK_STREAM
sock = socket(AF_INET,SOCK_STREAM)
sock.connect((argv[1],int(argv[2])))
print 'sock=', sock
while 1:
print 'enter something:'
msg = stdin.readline()
if msg:
sock.sendall(msg.strip()) # strip nl
else:
break
7.3 Parameter Studies
One straightforward application of task parallelism is the "parameter study", in which the same sequential program is run multiple times with different sets of input parameters. Since the program to be run is sequential, there is no communication among the workers, and the manager can be a simple script that communicates with the workers by means of the arguments to the sequential program and its standard output. We can start the workers with ssh and collect the output by using the popen system call, which returns a file descriptor we can select on and read the remote process's stdout from.
Although both the algorithm we use and its implementation are general, we present here a concrete example. We explore the parameter space of compiler options for the default Linux C compiler gcc. The man page for gcc conveniently lists in one place all the options that can be passed to gcc to cause it to produce faster machine code. Here is an excerpt from the man page:
For the matrix-matrix multiply program we are going to test with, which has no function calls, only some of these look useful. Here is a subset of the above list containing optimization flags that might have an effect on the speed of the program:
Since there are twelve switches that can be either present or absent, there are 212 possible combinations. These are not completely independent, since some switch settings imply others, especially the three -0 flags, but we will ignore thus fact for the sake of simplifying our example, and just try all 4096 combinations, Indeed, which switch settings are redundant in the presence of others should be deducible from our results!
Our plan will be to take a simple test program and compile it with all possible switch combinations and run it, reporting back the times. Since we have 4096 jobs to run, the use of a cluster will make a big difference, even if the individual tasks are short.
For our test program, we will use a straightforward matrix-matrix multiply program, shown in
Figure 7.10. It multiples two 300 ?300 matrices, timing the calculation, this may not be the highest performing way to do this, but it will do for our purposes. The program echoes its command line arguments, which it does not otherwise use; we will use them to help us record the arguments used to compile the program.
Our worker programs will just be the sequence
and the manager will start them with ssh, on hosts whose names are in a file. The other argument to our manager is a file of possible arguments. It contains exactly the twelve lines listed above. The manager just steps through the numbers from 0 up to the total number of runs (in our case 4096) treating each number as a binary number where a 1 bit represent the presence of the compiler switch corresponding to that position in the binary number. Thus we will run through all possible combinations.
The overall plan is to loop through the parameter space represented by the binary numbers represented by the binary numbers from 0 to 212. If there is a free host (no worker is working there) we assign it the next task; if not we select on the sockets that are open to currently working workers. When one of them reports back, we add it back to the list of free hosts. At the end, after all the work has been assigned, we still have to wait for the last tasks to complete.
Let us step through the code in Figure 7.11 in detail. First we read in the list of hosts (initial value of the list freeHosts) and the list of possible arguments (parmList). We initialize the set of sockets to select on to empty since there are no workers yet, and create an empty Python dictionary (fd2host) where we will keep track of busy hosts and the connections to them. We set numParmSets to the number of subtasks, which we can calculate from the number of possible compiler flags in the input file. Then we enter the main loop, which runs until we have assigned all the work and there are no outstanding workers working. If there is a subtask still to do and a free host to do it on, we construct the parameter list corresponding to the next task (in ParmSet), and pick the first host from the list of free hosts, temporarily removing it from the list. We then build a string containing the specification of the subtask. The Popen3 command forks a process that runs the ssh program locally, which runs the gcc-matmult sequence remotely. The ssh's, and therefore the remote processes, run in parallel.
We set runfd to the stdout of the ssh, which collects the stdout from the matmult. Each line of stdout will contain the time followed by the compiler flags used. Then we add this fd to the list of sockets available for selecting on and enter into the list fd2host the host attached to that fd.
If there are no free hosts or we have already assigned all the subtasks, then we select on the sockets connected to busy workers. When one those sockets becomes active, it means that the associated worker has set us a line of output. We read it and print it, or if the read fails (the worker exited, which sends an EOF on the socket), we close that socket, take it out of the list of sockets to select on, and add the corresponding host to the list of free hosts, since it can mow be assigned another subtask.
The manager exits once all the subtasks have been done and all the workers have completed. If we run this with
then we will get the best combinations at the top of the list. Try it!
7.5 Decomposing Programs Into Communicating ProcessesNot all problems can be divided into independent tasks. As we saw in
Section 1.3.6, some applications are too large, in terms of their memory or compute needs, for a single processor or even a single SMP node. Solving these problems requires breaking the task into communicating (rather than independent) processes. In this section we will introduce two examples of decomposing a single task into multiple communicating processes. Because these programs are usually written using a message-passing programming model such as MPI or PVM, the details of implementing these examples are left to the chapters on these programming models.
7.5.1 Domain Decomposition
Many problems, such as the 3-dimensional partial differential equation (PDE) introduced in
Section 1.3.6, are described by an approximation on a mesh of values. This mesh can be structured (also called regular) or unstructured. These meshes can be very large (as in the example in
Chapter 1) and require more memory and computer power than a single processor or node can supply. Fortunately, the
For simplicity, we consider a two-dimensional example. A simple PDE is the Poisson equation,
where f is a given function and the problem is to find u. To further simplify the problem, we have chosen Dirichlet boundary conditions, which just means that the value of u along the boundary is zero. Finally, the domain is the unit square [0, 1] ?[0, 1]. A very simple discretization of this problem uses a finite difference approximation to the derivatives, yielding the approximation
Defining a mesh of points (xi, yj) = (i ?h, j ?h) with h = 1/n, and using the ui,j to represent the approximation of u(xi, yj), we get
| (7.1) | 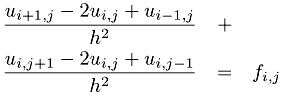 |
We can now represent this using two dimensional arrays. We'll use Fortran because Fortran has some features that will make these examples easier to write. We will use U(i, j) as our computed value for ui,j.
To solve this approximation for the Poisson problem, we need to find the the values of U. This is harder than it may seem at first, because
Equation 7.1 must be satisified at all points on the mesh (i.e., all values of i and j) simultaneously. In fact, this equation leads to a system of simultaneous linear equations. Excellent software exists to solve this problem (see
Chapter 12), but we will use a very simple approach to illustrate how this problem can be parallelized. The first step is to write this problem as an iterative process
This is the Jacobi iteration, and can be written in Fortran as
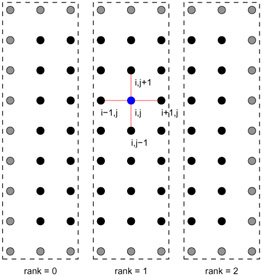
At this point, we can see how to divide this problem across multiple processors. The simplest approach is to divide the mesh into small pieces, giving each piece to a separate processor. For example, we could divide the original mesh (U(0:n,0:n) in the code) into two parts: U(0:n,0:n/2) and (U(0:n,n/2+1:n). This approach is called domain decomposition, and is based on using the decompositions of the physical domain (the unit square in this case) to create parallelism.
Applying this approach for two processors, we have the two code fragments shown in
Figure 7.12. Note that each process now has only half of the data because each array is declared with only the data "owned" by that processor. This also shows why we used Fortran; the ability to specify the range of the indices for the arrays in Fortran makes these codes very easy to write.
However, unlike the decompositions into independent tasks in the first part of this chapter, this decomposition does not produce indepentent tasks. Consider the case of j=n/2 in the original code. Process zero in
Figure 7.12 computes the values of UNEW(i,n/2). However, to do this, it needs the values of U(i,n/2+1). This data is owned by processor one. In order to make this code work, we must communicate the data owned by processor one (the values of U(i,n/2+1) for i=1,...,n-1) to processor zero. We must also allocate another row of storage to hold these values; this extra row is often called a ghost points or a halo. The resulting code is shown in
Figure 7.13.
Note also that although both processes have variables named UNEW and i, these are different variables. This kind of parallel programming model is sometimes called a shared-nothing model because no data (variables or instructions) are shared between the processes. Instead, explicit communication is required to move data from one process to another.
Section 8.3 discusses this example in detail, using the Message Passing Interface (MPI) to implement the communication of data between the processors, using code written in C.
There are more complex and powerful domain decomposition techniques, but they all start from dividing the domain (usually the physical domain of the problem) into a number of separate pieces. These pieces must communicate along their edges at each step of the computation. As described in
Section 1.3.6, a decomposition into squares (in two-dimensions) or cubes (in three dimensions) reduces the amount of data that must be communicated because those shapes maximize the volume to surface area ratio for rectangular solids.
7.5.2 Data Structure Decomposition
Not all problems have an obvious decomposition in terms of a physical domain. For these problems, a related approach that decomposes the data-structures in the application can be applied. An example of this kind of application is the solution of a system of linear equations Ax = b, where the equations are said to be dense. This simply means that most of the elements of the matrix describing the problem are non-zero. A good algorihm for solving this problem is called LU factorization, because it involves first computing a lower trianular matrix L and an upper triangular matrix U such that the original matrix A is given by the product LU. Because an lower (resp. upper) triangular matrix has only zero elements below (resp. above) the diagonal, it is easy to find the solution x once L and U are known. This is the algorithm used in the LINPACK
[34] benchmark. A parallel verison of this is used in the High-Performance Linpack benchmark, and this section will sketch out some of the steps used in parallelizing this kind of problem.
The LU factorization algorithm looks something like the code shown in
Figure 7.14, an n ?n matrix A represented by the Fortran array a(n,n).
An obvious way to decompose this problem, following the domain decomposition discussion, is to divide the matrix into groups of rows (or groups of columns):
However, this will yield an inefficient program. Because of the outer-loop over the rows of the matrix (the loop over i), once i reaches n/4 in the case of four processors, processor zero has no work left to do. As the computation proceeds, fewer and fewer processors can help with the computation. For this reason, more complex decompositions are used. For example, the ScaLAPACK library uses the two-dimensional block-cyclic distribution shown here:
This decomposition ensures that most processors are in use until the very end of the algorithm.
Just as in the domain decomposition example, communication is required to move data from one processor to another. In this example, data from the ith row must be communicated from the processors that hold that data to the processors holding the data needed for the computations (the loops over j). We do not show the communication here; see the literature on solving dense linear systems in parallel for details on these algorithms.
The technique of dividing the data structure among processors is a general one. Chosing the decomposition to use requires balancing the issues of load balance, communication, and algorithm complexity. Addressing these may suggest algorithmic modifications to provide better parallel performance. For example, certain variations of the LU factorization method described above may perform the floating-point operations in a different order. Because floating-point arithmetic is not associative, small differences in the results may occur. Other variations may produce answers that are equally valid as approximations but give results that are slightly different. Care must be exercised here, however, because some approximations are better behaved than others. Before changing the algorithm, make sure that you understand the consequences of any change. Consult with a numerical analysist or read about stability and well-posedness in any textbook on numerical computing.
7.5.3 Other Approaches
There are many techniques for creating parallel algorithms. Most involve dividing the problem into separate tasks that may need to communicate. For an effective decomposition for a Beowulf cluster, the amount of computation must be large relative to the amount of communication. Examples of these kinds of problems include sophisticated search and planning algorithms, where the results of some tests are used to speed up other tests (for example, a computation may discover that a subproblem has already been solved.).
Some computations are implemented as master/worker applications, where each worker is itself a parallel program (e.g., because of the memory needs or the requirement that the computation finish within a certain amount of time, such as overnight). Master/worker algorithms can also be made more sophisticated, guiding the choice and order of worker tasks by previous results returned by the workers.
Chapter 8: Parallel Programming with MPI
Overview
William Gropp and Ewing Lusk
Chapter 7 described how parallel computation on a Beowulf is accomplished by dividing a computation into parts, making use of multiple processes and executing each on a separate processor. Sometimes an ordinary program can be used by all the processes, but with distinct input files or parameters. In such a situation, no communication occurs among the separate tasks. When the power of a parallel computer is needed to attack a large problem with a more complex structure, however, such communication is necessary.
One of the most straightforward approaches to communication is to have the processes coordinate their activities by sending and receiving messages, much as a group of people might cooperate to perform a complex task. This approach to achieving parallelism is called
message passing.
In this chapter and the next, we show how to write parallel programs using MPI, the Message Passing Interface. MPI is a message-passing library specification. All three parts of the following description are significant.
-
MPI addresses the message-passing model of parallel computation, in which processes with separate address spaces synchronize with one another and move data from the address space of one process to that of another by sending and receiving messages. [
1]
-
MPI specifies a library interface, that is, a collection of subroutines and their arguments. It is not a language; rather, MPI routines are called from programs written in conventional languages such as Fortran, C, and C++.
-
MPI is a specification, not a particular implementation. The specification was created by the MPI Forum, a group of parallel computer vendors, computer scientists, and users who came together to cooperatively work out a community standard. The first phase of meetings resulted in a release of the standard in 1994 that is sometimes referred to as MPI-1. Once the standard was implemented and in wide use a second series of meetings resulted in a set of extensions, referred to as MPI-2. MPI refers to both MPI-1 and MPI-2.
As a specification, MPI is defined by a standards document, the way C, Fortran, or POSIX are defined. The MPI standards documents are available at
www.mpi-forum.org and may be freely downloaded. The MPI-1 and MPI-2 standards are available as journal issues [
72, 73] and in annotated form as books in this series [
105, 46]. Implementations of MPI are available for almost all parallel computers, from clusters to the largest and most powerful parallel computers in the world. In
Section 8.9 we summarizes the most popular cluster implementations.
A goal of the MPI Forum was to create a powerful, flexible library that could be implemented efficiently on the largest computers and provide a tool to attack the most difficult problems in parallel computing. It does not always do the simplest tasks in the simplest way but comes into its own as more complex functionality is needed. As a result, many tools and libraries have been built on top of MPI (see
Table 9.1 and Chapter 12). To get the flavor of MPI programming, in this chapter and the next we work through a set of examples, starting with the simplest.
|
|

 LINUX
LINUX 
 Books
Books